A dwarf planet designated 2015 RR245 (and now in search of a name) has been found in an orbit that takes it out to at least 120 AU. It’s a discovery made by the Outer Solar System Origins Survey (OSSOS), an international collaboration focused on the Solar System beyond Neptune. The goal is to test models of how the Solar System developed by studying the movements of icy objects, many of which may have been destroyed or ejected from the Solar System altogether through movements of the giant planets early in the formation process.
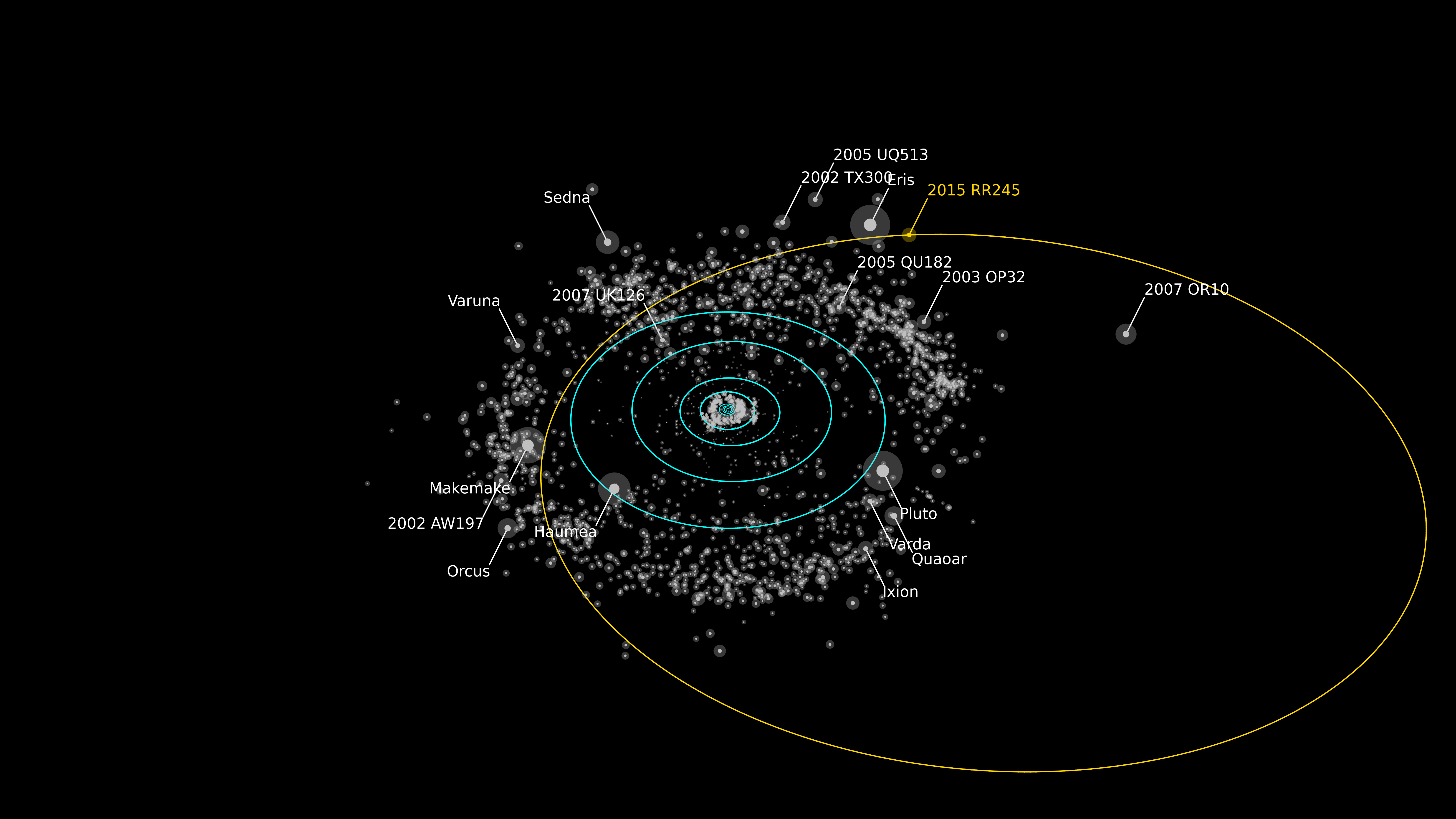
Image: Rendering of the orbit of RR245 (orange line). Objects as bright or brighter than RR245 are labeled. The blue circles show the projected orbits of the major planets. The Minor Planet Center describes the object as the 18th largest in the Kuiper Belt. Credit: Alex Parker/OSSOS team.
We’ve had a close look at one dwarf planet at the edge of the system when New Horizons flew past Pluto a year ago, and with a diameter of roughly 700 kilometers, 2015 RR245 will itself be worthy of future investigation. We now know from the New Horizons example that distant icy worlds like these produce exotic landscapes and unexpected geological processes. But their orbits may tell us just as much as their surface features. Consider:
Perihelion for 2015 RR245 will take place toward the end of this century, though refining its orbit will take time. According to the project’s website, OSSOS can work out TNO orbits in about 16 months on average, which is actually less than 1% of the time it takes them to orbit the Sun. While thinking about these orbits, bear in mind that although the number of TNOs so far discovered is growing, we’re surely seeing a small subset of what must be a vast population.
We’re tracking, in other words, only those objects that are relatively close to the Sun and thus are the easiest to spot. Michele Bannister (University of Victoria BC) touches on the matter in this news release:
“The icy worlds beyond Neptune trace how the giant planets formed and then moved out from the Sun. They let us piece together the history of our Solar System. But almost all of these icy worlds are painfully small and faint: it’s really exciting to find one that’s large and bright enough that we can study it in detail.”
But the fact that we find so few TNOs that are large and bright enough for such study is itself an issue. The second Kuiper Belt Object found (1992 QB1) was discovered over sixty years after the first (Pluto), an indication of how small and faint these objects are. Ethan Siegel speaks to this in an essay for Forbes, noting that objects like 2015 RR245 have extremely eccentric orbits, in this case one with perihelion at roughly 34 AU and aphelion at 120 AU.

We’re reminded that we have not just Kuiper Belt Objects to contend with but scattered disk objects as well (scattered disk objects are considered to be KBOs with large orbital eccentricities), and beyond them the so-called Sednoids. This has implications for the conjectured Planet Nine, whose existence has been inferred through the clustering of a small number of Sednoid objects. Siegel has his doubts, and they’re reinforced by 2015 RR245:
But it’s also possible, as scattered disk objects and elliptical KBOs show, that there are a huge variety of objects with tremendously varied orbits out there, and we’re only seeing a tiny fraction of them. If the objects we’re seeing have even a slight bias to them, it could lead us to jump to all sorts of incorrect conclusions, just as we did decades ago claiming periodic mass extinctions due to asteroid impacts and the Nemesis theory of a second Sun. Incomplete data is what we’ve got, and the first results of OSSOS and the discovery of 2015 RR245 should remind us all of how much more there is — not just in the Universe but even in our Solar System — still left to discover.
Image: Discovery images of RR245. The images show RR245’s slow motion across the sky over three hours. Credit OSSOS team.
Meanwhile, OSSOS plugs away, having already discovered more than five hundred new trans-Neptunian objects. 2015 RR245 is its largest discovery and the only dwarf planet found by the team, which uses the MegaPrime camera, an imager on the 3.6m Canada-France-Hawaii Telescope (CFHT). As larger telescopes like the Large Synoptic Survey Telescope (LSST) come online capable of mapping the entire visible sky in just a few nights, we will doubtless gain a better idea of the actual distribution of these varied objects. That may put to bed once and for all the question of whether a large undiscovered planet is out there at system’s edge.



Talking of Planet Nine, a paper appeared on the arXiv today investigating whether such a planet could explain the misalignment between the sun’s rotation and the orbital plane of the planets.
Bailey, Batygin & Brown (arXiv:1607.03963 [astro-ph.EP]) “Solar Obliquity Induced by Planet Nine”
From the abstract:
Could this also of been done by Planet 9 being kicked out of the inner solar system? Or even more likely it was slowly pumped out!
Another paper about Planet Nine and the inclination of the known solar system planets:
Gomes, Deienno and Morbidelli (arXiv:1607.05111 [astro-ph.EP]) “The inclination of the planetary system relative to the solar equator may be explained by the presence of Planet 9“
It is unlikely due to the possible planet 9 because the mass of that planet is expected to be less than Neptune in mass. Misalignments in planetary systems is quite common, they can get quite chaotic in the final phases of planetary formation.
I see what you mean:
Spin-orbit Misalignment as a Driver of the Kepler Dichotomy:
Christopher Spalding, Konstantin Batygin
http://arxiv.org/abs/1607.03999
Dynamics of Stellar Spin Driven by Planets Undergoing Lidov-Kozai Migration: Paths to Spin-Orbit Misalignment:
Natalia I. Storch, Dong Lai, Kassandra R. Anderson
http://arxiv.org/abs/1607.03937
So — lay question here — would it help materially to have a space telescope either stationed or at least on a receding trajectory “above” or “below” the solar system? In order to more easily isolate these deep solar system objects and determine their orbits.
Perhaps one of those “above” or “below” trajectories also would be optimum in terms of having the least clutter from non-solar system objects in the background behind the target deep solar system objects. Such an optimum more-background-clutter-free trajectory perhaps might be more or less oblique rather than necessarily strictly orthogonal to the plane of the ecliptic.
If so, a sundiver type mission profile might work well in setting the telescope on the desired trajectory.
Also might be interesting to study our system from that perspective from greater and greater distances — including studying the Sun’s wobble — for comparison to our views of other more distant systems as to which we have a similar directional perspective. We know the constituents of our system — at least close in — fairly well, which then might allow for a more meaningful comparison between views of our Sun’s wobble at a distance and our view of other stars.
The authors are quite right in suggesting that the evidence for Planet 9 may be due to observation bias. This is hard to rule out, and I am not convinced that it has been, sufficiently. So, until it is actually discovered, I am not counting on there being a Planet 9.
This just in:
Warm Jupiters are less lonely than hot Jupiters:
https://www.sciencedaily.com/releases/2016/07/160714110916.htm
http://iopscience.iop.org/article/10.3847/0004-637X/825/2/98/meta;jsessionid=EFFE4E417FE8676C35510FAA34E90AB5.c1.iopscience.cld.iop.org
The central idea is that there are 2 kinds of planetary systems with warm Jupiters: those that formed in situ, which have additional planets; and those that formed ex situ, followed by inward migration. These don’t have any additional planets, similar to the hot Jupiter systems. The distribution would be about half-half.
I have a question that does not directly relate to the topic discussed here. Where would there be a probable, reasonably accessible resource of helium-3? We’d need it. I am thinking of a ‘planet nine’, being a resonably low gravity well very far away from the Sun as being a possible candidate. I understand the Moon has some, bound up in regolith, but I have difficulty envisaging ‘mining’ the quantities neede some centuries down the road(?). We need to look for such a resource. I have this notion it needs to be ver far away, a really distant, yet ‘reachable’, not too big planet being something to look for.
The Moon has reasonable amounts of 3He in its regolith, but in very low concentrations, therefore extracting usable amounts of 3He means scraping huge portions of the lunar surface. Gas giants have probably good concentrations of 3He (as He is one of their main constituents). Even though mining Jupiter might not be practical due to its deep gravity well, it might be easier to mine Saturn, Uranus and Neptune with some kind of balloon or spacecraft skimming their atmospheres.
Perhaps a laser in orbit or on a balloon could be used to ionise the he3 selectively and then a powerful magnetic field carrying craft in orbit is used to catch it.
Why do you want Helium-3? Other than for sightly heavier party balloons and some medical uses, it’s nothing special.
IF a nuclear fusion reactor technology that requires He-3 is ever developed, the easiest way of getting He-3 is to produce it on the ground, as the byproduct of Tritium decay. It would be expensive, but a lot cheaper and easier than building the technology and infrastructure to mine it elsewhere in the solar system and return it safely to the Earth.
If there is going to be a practical means of interplanetary travel in the outer Solar System, or to even more faraway places than that, there will be a need for some kind of a reaction engine with a very high specific impulse. And if the transit times are going to be shorter than significant parts of human lifespans, there is a need for something with a reasonably high trust to weight ratio. We have seen some of the propulsion concepts being considered, but one out of very few concepts that offer a reasonable trust to weight ratio combined with a very high specific impulse, and that is also in some kind of development, is a pulsed-fusion concept called magneto-inertial confinment.
A magneto inertial confinement reaction engine can, to my knowlegde, run on two different kinds of fuel. 1) deuturium-tritium, and, 2), helium-3. The deuturium tritium fuel is the most available of the two, but produces intense neutron radiation when burnt. It hurts the structure surrounding the reaction zone, limiting engine life. It is also unstable, as tritium is radioactive. If a deep space infrastructure is built with this technology embedded, this will be the fuel used to get underway. Helium-3 is difficult to obtain. But it is stable, and it burns ‘cleaner’, in that it does not produce the neutron flux, letting the engine structure last longer. It also has a higher specific impulse than the deuturium-tritium fuel, and is the fuel of choice, particularly in applications where demands are extreme, say, a star probe. If fuel can be obtained.
And of course, it would be useful in party balloons. Actually, it is lighter than conventional helium, so He 3 party balloons should fly better – honestly speaking, I haven’t tested that one. One more reason to go find a good source.
Magnetic inertial confinemenet technology is being developed, and will be available if the right people get the resources and the peace and the quiet to do the job.
NASA is currently sponsoring a study considering a fusion driven probe that would land on Pluto: https://www.nasa.gov/feature/fusion-enabled-pluto-orbiter-and-lander As far as I get it, this is not magneto inertial confinement, but a related consept that is part of the Princeton fusion research program.
Some time back, NASA sponsored a study featuring a fusion powered interplanetary spaceship for bringing astronauts to the Jovian system: http://ntrs.nasa.gov/archive/nasa/casi.ntrs.nasa.gov/20040010797.pdf
And this technology is being developed, among others, by this company, and I believe NASA is involved with some financing here also:http://msnwllc.com/our-mission
Fusion has proved to be a difficult thing to do, but it is coming, even if it has taken time. And we need a good source of helium-3.
Yes, I can see that. But as evidence is mounting there are more worlds lurking in the distant parts of the Solar System, I am intrigued by the prospect of finding really distant planets that sport atmospheres of only hydrogen/helium, or ones that may even have a helium ocean sloshing around, and maybe at a size of only a fraction of, say, Neptune. Getting the stuff into orbit around a small world would not be such a daunting task. Hence this question right in the middle of a blog discussing dwarf planets.
A continual lesson for SETI: When searching for one thing in astronomy, one always finds something else along the way….
https://carnegiescience.edu/node/2082